Cómo el INVIMA pudiese agilizar el trámite para la aprobación de protocolos de investigación clínica con dispositivos médicos y así unir a Colombia al ecosistema global de innovación médica
Colombia tiene la oportunidad histórica de convertirse en la estrella mundial de la investigación clínica con tecnologías médicas innovadoras. El país necesita un INVIMA moderno, ágil y predecible que apruebe nuevos estudios en máximo 30 días.

Según Wikipedia, "Un ensayo clínico es una evaluación experimental de un producto, sustancia, medicamento, técnica diagnóstica o terapéutica que, en su aplicación a seres humanos, pretende valorar su eficacia y seguridad". Los estudios de prometedores tratamientos nuevos o experimentales en pacientes se conocen cómo ensayos (comúnmente llamados "estudios") clínicos. Un estudio clínico se realiza sólo cuando hay razones para creer que el tratamiento que se está estudiando puede ser beneficioso para el paciente.
El país debe fomentar un ambiente regulatorio ágil, predecible y eficiente para atraer del extranjero cada vez más estudios de investigación clínica con diferentes innovaciones médicas (medicamentos farmacéuticos y biológicos, biotecnología, dispositivos médicos, terapia génica, terapia celular, ingeniería de tejidos, etc.). Los actuales indicadores de rezago en el número de estudios de investigación clínica que se realizan en el país, presentan una oportunidad histórica para que Colombia haga los cambios normativos y administrativos necesarios para potencializar su industria de investigación clínica.
Colombia tiene todo para convertirse en una potencia no solo regional sino mundial en el desarrollo de estudios clínicos. Esa es una de las principales conclusiones de un análisis que desarrolló recientemente la firma de investigaciones Pugatch Consilium. El análisis estima que Colombia podría generar hasta USD 500 millones en inversión extranjera directa si se hacen los cambios necesarios en el país. Leer más.
Las empresas que desarrollan nuevas tecnologías y dispositivos médicos son generalmente extranjeras (mayormente provenientes de 10 estados en EE. UU.) y buscan centros de investigación en clínicas y hospitales en Colombia y otros países de Latinoamérica para realizar los estudios de investigación clínica necesarios para que la FDA u otras entidades regulatorias aprueben la comercialización de sus dispositivos médicos. El artículo titulado Should You Conduct Your Medical Device Clinical Trial In Latin America?, también escrito por el autor de esta columna, Julio G. Martinez-Clark, ilustra el crecimiento de los estudios clínicos con dispositivos médicos en Latinoamérica.
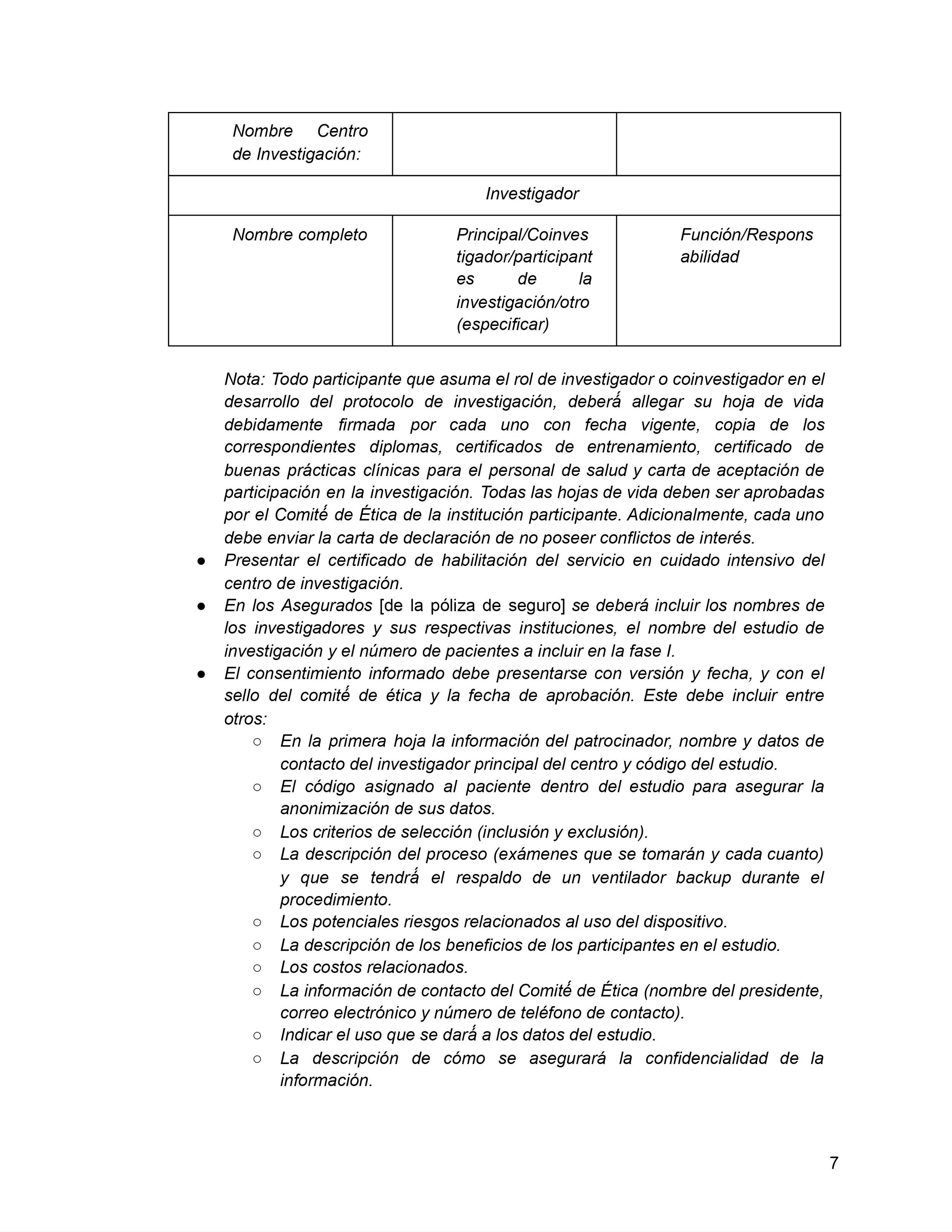
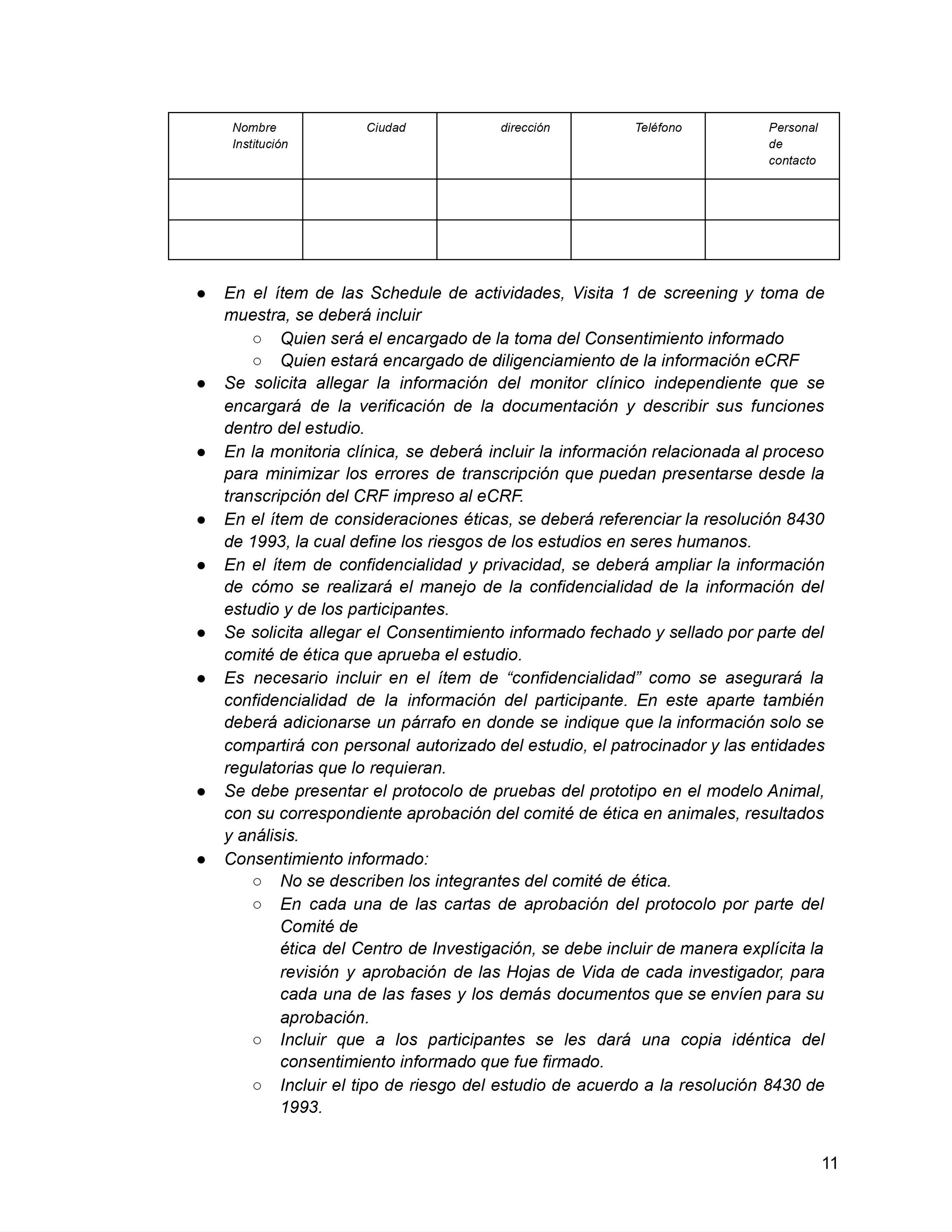
En Colombia, el trámite de aprobación de un protocolo de investigación clínica con un nuevo producto médico (eje. dispositivo médico o una nueva molécula farmacéutica) involucra tres pasos:
Aprobación del comité de ética en investigación (CEI) correspondiente a la institución prestadora de servicios de salud (I.P.S.) dónde se realizará el estudio;
Aprobación ante el Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA); y
Aprobación del registro de importación ante la Ventanilla Única de Comercio Exterior (VUCE) del Ministerio de Comercio, Industria y Turismo (MinCIT).
A continuación exponemos dos ejemplos de cómo el trámite de aprobación de un protocolo de investigación clínica con un dispositivo médico ante la Sala de Dispositivos Médicos y Reactivos de Diagnóstico In Vitro del INVIMA (la “Sala”) es un proceso impredecible, ineficiente y le resta agilidad y competitividad al país. También resumiré los obstáculos que hemos encontrado al solicitar la aprobación de estudios de investigación clínica ante la Sala, mencionaré los tiempos de aprobación de un estudio de investigación clínica en otros países, y sugeriré unas oportunidades de mejora al trámite de aprobación de un estudio de investigación clínica en Colombia.
Ejemplos que han hecho que empresas extranjeras desistan de realizar sus estudios de investigación clínica en Colombia.
En el caso específico de uno de nuestros clientes, CeloNova, la Sala aprobó el protocolo COBRA en el Acta 08 de 2014 y luego lo desaprobó en el Acta 10 de 2014 alegando que faltaba la carta de aprobación del Comité de Ética del centro de investigación Angiografía de Occidente en Cali (esta carta se había entregado en los documentos originales y sin esta nunca se hubiese aprobado el protocolo en el Acta 08 de 2014). En el item 3.14 del Acta 7 de 2014 la Sala solicitó cartas firmadas por los investigadores que nunca antes había solicitado y que no hacen parte de la lista oficial de requisitos para aprobación de protocolo de investigación publicada por la Sala como se puede ver aquí.
La Sala también confundió los documentos del estudio COMBINE (del patrocinador Avinger) con los documentos del estudio COBRA (del patrocinador CeloNova). La radicación inicial de la solicitud de aprobación del protocolo COBRA se hizo el 25 de abril de 2014 y solo hasta el 20 de noviembre de 2014 la Sala publicó el acta de aprobación del protocolo (ver Acta 11 de 2014); 7 meses después.
En vista del prolongado tiempo de aprobación del protocolo COBRA, el patrocinador CeloNova decidió cancelar su estudio clínico en Colombia y la I.P.S. donde se planeaba conducir el estudio dejó de exportar casi USD $500 mil, sin contar con los otros beneficios intangibles (intercambio de conocimiento entre investigadores nacionales y extranjeros, la posibilidad de que los investigadores nacionales publiquen los resultados del estudio en revistas internacionales, etc.) —pero no menos importantes— que este estudio le hubiese traído al país.
Obstáculos que hemos observado al solicitar la aprobación de estudios de investigación clínica ante la Sala
A continuación resumimos los obstáculos que hemos observado al solicitar la aprobación de estudios de investigación clínica con dispositivos médicos prototipos ante la Sala:
El subir los documentos en formato .pdf a la plataforma del INVIMA es laborioso y confuso. La plataforma tiene un límite de tamaño de archivo y para un dossier de casi 1 000 páginas toca dividirlo en decenas de archivos pequeños.
La Sala tiene una lista de requisitos para la aprobación de un estudio de investigación clínica con dispositivos médicos y proporciona un documento llamado Formato De Presentación Y Evaluación Protocolos De Investigación Clínica Con Dispositivos Médicos Y Otras Tecnologías. Este documento no contiene la lista completa de requisitos para la aprobación de un estudio clínico. En varias Actas, la Sala ha solicitado requisitos diferentes o adicionales a los contemplados en este formato. Esto hace que el solicitante no tenga claridad sobre la lista de requisitos que la Sala le exige y le resta predecibilidad al proceso de aprobación de un estudio de investigación clínica. En el caso de nuestra empresa, bioaccess.™, leemos cada una de las actas que emite la Sala después de la reuniones de la Comisión Revisora y hemos compilado nuestra propia lista extra-oficial de requisitos para la aprobación de un estudio de investigación clínica con dispositivos médicos.
Una de las observaciones en el Formato De Presentación Y Evaluación Protocolos De Investigación Clínica Con Dispositivos Médicos Y Otras Tecnologías (Código: ASS-RSA-FM085. Versión: 01. Fecha de Emisión: 08/03/2018) es “La información de la literatura científica debe ser suministrada en el idioma del país de origen con una traducción al idioma castellano”. El número total de páginas en los documentos que exige la Sala puede fácilmente alcanzar 1 000 páginas. El exigirle al solicitante que traduzca toda la literatura científica al idioma castellano le agrega tiempo y costo adicional al proceso de preparación para una solicitud de aprobación de un estudio clínico. Esto le resta competitividad al país debido a que otros países (eje. COFEPRIS en México) aceptan esta documentación científica en inglés. Sería ideal que la Sala permitiera entregar la documentación científica en inglés y que le permitiera al solicitante entregar el resumen del protocolo de investigación en idioma castellano.
Al aprobar un protocolo de investigación, la Sala no usa un formato claro al aprobar un protocolo de investigación clínica. La Sala publica un acta de cada una de sus sesiones y este documento no es una copia controlada ni necesariamente valida y entendible al solicitar el respectivo Registro de Importación ante la Ventanilla Única de Comercio Exterior (VUCE) del MinCIT para que los dispositivos médicos en investigación puedan ser importados al país y usados en el estudio en la I.P.S. donde se conducirá el proyecto de investigación.
El documento de aprobación del INVIMA, no cumple siempre con los requerimientos que exige el VUCE; esto hace difícil la apertura de un registro de importación de los dispositivos que serán usados en el protocolo. Por ejemplo, no siempre se menciona claramente la persona jurídica que la Sala autoriza para la importación, y no siempre se menciona el número exacto y número de modelo/lote de dispositivos médicos que la Sala autoriza importar. Esto hace que el VUCE rechace las solicitudes de Registro de Importación y fuerza al solicitante a pedirle a la Sala una aclaración del acta donde aprobó el estudio; esto le genera al solicitante retrasos en comenzar su proyecto de investigación.
La Sala fuerza a que el solicitante realice otro trámite en serie ante el VUCE. Debido a que la solicitud del registro de importación es considerado un proceso aduanero previo a que la empresa extranjera que patrocina el estudio despache los productos a ser nacionalizados en Colombia, este trámite es generalmente realizado por una agencia de aduanas, lo cual conlleva a involucrar a un tercero más en el proceso con sus correspondientes costos y retrasos.
La publicación de las actas de la Sala toma alrededor de 15 días hábiles. Después de cada sesión de la Sala, esta publica en la página web del INVIMA sus decisiones en un acta; esta es publicada generalmente 15 días hábiles después del día de la sesión. 15 días es demasiado tiempo para saber cuál fue la decisión de la Sala.
Largo periodo de respuesta del INVIMA. Un solicitante radica su documentación antes de la fecha de corte publicada por el INVIMA y tiene que esperar aproximadamente 30 días (más los 15 días que toma en publicarse el acta de sesión de la Sala) hasta que ocurra la sesión ordinaria de la Comisión Revisora para obtener retroalimentación de su solicitud. Dado el hecho que la Sala publica el acta de la sesiones ordinarias aproximadamente de 7 a 10 días después de la fecha de una sesión ordinaria, el solicitante no tiene tiempo de obtener la documentación que se le requirió en la sesión anterior ya que muchas veces la fecha de corte de la próxima sesión ordinaria ya ha pasado o está a solo unos días de pasar; esto fuerza al solicitante a esperar aproximadamente otros 30 días hasta la siguiente fecha de reunión de la Sala. Ante esta situación, un solicitante pierde aproximadamente 60 días en total desde la fecha de una sesión ordinaria en la cual se le revisa por primera vez su solicitud y hasta que se le revise la documentación que entregó como requisito en esa primera sesión.
La publicación de la totalidad de las actas en el sitio web del INVIMA como resultado de las sesiones ordinarias de la Sala, revela información confidencial y competitiva de los solicitantes, lo cual no siempre protege la propiedad intelectual de los mismos.
Las actas que se crean como resultado de cada sesión de la Sala se convierten en la única prueba legal de que un protocolo se ha aprobado. Estas se publican en formato PDF en la página web del INVIMA e incluyen nombres, apellidos y cargos de funcionarios, sin embargo no figuran firmas de ninguno de ellos y esto pone en duda la validez legal de estas actas al frente a las distintas entidades y personas a las cuales se les presenta como prueba de aprobación (fabricantes extranjeros patrocinadores de los estudios, sociedades de intermediación aduanera, personas naturales y jurídicas en Colombia, etc.).
En varias ocasiones a los miembros de la Sala no les ha alcanzado el tiempo para cubrir todos los temas a tratar en una sesión ordinaria (de 8:00 a.m. a 5:00 p.m.). Cuando un solicitante entrega su documentación antes de la fecha de corte para una sesión ordinaria, espera que su solicitud sea revisada en la próxima sesión ordinaria (programada aproximadamente 30 días después); al no revisarse su solicitud en en esa sesión ordinaria, tiene que esperar aproximadamente otros 30 días hasta la próxima sesión ordinaria —sin garantía de que su solicitud sea revisada durante esa siguiente sesión. Esto adiciona un factor de impredecibilidad al trámite y le resta competitividad al país.
En varias ocasiones la Sala ha negado protocolos por supuesta falta de un documento que fue adjuntado en la radicación original de la solicitud de aprobación del protocolo.
En varias ocasiones la Sala ha confundido documentos de un protocolo con otro.
El INVIMA y los CEI parecen tener papeles redundantes. De acuerdo a el Artículo 25 del Acuerdo No. 003 de 2014 del INVIMA, la función de la Sala es "conceptuar técnicamente sobre los protocolos de investigación de dispositivos médicos y otras tecnologías, que requieran autorización del INVIMA para su ejecución". La página 9 del documento del INVIMA titulado ABC Guía Comité de Ética en Investigación define las responsabilidades de un CEI en una I.P.S. Los conceptos de la Sala, sugieren que las funciones de la misma son redundantes a las funciones de los CEI que el mismo INVIMA certifica, regula y vigila. Por lo tanto, muchos aspectos de la evaluación de un protocolo de investigación clínica son duplicados —primero los evalúa técnicamente el CEI y los mismos aspectos luego son re-evaluados por el INVIMA. Bajo este actuar del INVIMA, no es claro cuál es la función "técnica" de la Sala: ¿la Sala evalúa protocolos de investigación con el fin de autorizar la importación de los productos médicos en investigación? ¿La Sala los evalúa con el fin de garantizar la seguridad de los pacientes, o los evalúa con ambos fines en mente? ¿Por qué la Sala evalúa la metodología del proyecto de investigación clínica? Es obvio pensar que la Sala evalúa protocolos con fines de aprobar la importación de los dispositivos médicos en investigación, pero no es obvia la razón por la cual la Sala evalúa técnica y científicamente un protocolo de investigación clínica para garantizar la seguridad de los pacientes cuando el mismo INVIMA ha autorizado la creación de un CEI en las I.P.S. del país para ejercer exactamente la misma función.
El trámite de aprobación de un estudio de investigación clínica es en serie, no en paralelo como en otros países. Es decir, INVIMA exige que el trámite se realice después de que el CEI de la I.P.S. donde se llevará el estudio apruebe el proyecto. Esto adiciona unos 30-60 días más al cronograma de un proyecto de investigación.
En cuanto a los tiempos de aprobación en otros países, para estudios de investigación clínica con dispositivos médicos, después de radicada la documentación e iniciado el trámite, la Sala rechaza o aprueba el proyecto en aproximadamente de 30 días (contados desde la fecha de corte de recibo de los documentos del trámite, hasta el día de la sesión de la Sala). Sin embargo, el INVIMA le informa indirectamente (con la publicación del acta de la sesión en la página del INVIMA) al solicitante sobre el rechazo o aprobación del estudio unos 15 días hábiles después de la sesión de la Sala. En esta página he creado una tabla que describe el tiempo del trámite de aprobación de un protocolo de investigación clínica en Colombia.














El 23 de octubre de 2015, las asociaciones gremiales del sector de investigación clínica en Colombia le envió una carta al MinCIT donde se le expone al Ministerio como el ineficiente trámite de aprobación de un protocolo de investigación clínica en el INVIMA le resta competitividad al país y hace que deje de exportar millones de dólares anuales en servicios de salud. En cuanto a la comparación del tiempo de aprobación del INVIMA con otros países, 30 días (en medicamentos y dispositivos) sería ideal para ser más competitivos internacionalmente.
Es importante ver el proceso de aprobación de un protocolo de investigación desde una perspectiva más amplia. Es mejor verlo desde la perspectiva del cliente final que deseamos atraer al país —el patrocinador del estudio (la empresa en EE.UU. que desea contratar los servicios de una I.P.S. en Colombia). Son varios los pasos que hay que seguir en un país antes de legalmente comenzar a reclutar pacientes en un estudio de investigación clínica; en esta tabla se observan los pasos típicos en un estudio clínico con sus correspondientes tiempos para el caso de Colombia.
Oportunidades de mejora en el trámite de aprobación de un estudio de investigación clínica con un dispositivo médico
Ante lo anterior, surge naturalmente la siguiente pregunta, a la cual le daré respuesta basado en nuestra experiencia y en la retroalimentación que hemos recibido de nuestros clientes (empresas extranjeras que diseñan y desarrollan tecnologías médicas): ¿cómo sería el trámite ideal ante el INVIMA para la aprobación de un estudio de investigación clínica con dispositivos médicos prototipo?
Que el INVIMA implemente un proceso predecible (una única lista de requisitos/documentos éticos, técnicos y legales que contenga todos los posibles documentos y requisitos al solicitante) con tiempos claros de aprobación (fecha de reuniones, fechas de publicación de actas o notificaciones electrónicas a los solicitantes).
Que el INVIMA publique guías que ayuden al solicitante a radicar sus solicitudes de evaluación (al igual que el INVIMA publicó para medicamentos):
Que el INVIMA permita radicar todos los documentos que son requisitos para la aprobación de un protocolo de investigación clínica en idioma inglés.
Que el INVIMA permita radicar fácilmente documentos (solicitudes de aprobación de protocolos y reporte de eventos adversos) de forma electrónica y le notifique al solicitante de forma electrónica sobre la aprobación de la solicitud o sobre la solicitud de documentación adicional.
Que el INVIMA establezca un mecanismo ágil de revisión previa de cada solicitud de aprobación de protocolo con el fin de darle la oportunidad a cada solicitante de corregir cualquier error en la documentación antes de la proxima reunión de la Comisión Revisora de la Sala.
Que si la Sala tiene una sesión y rechaza una solicitud por falta de documentación o aclaración, le permita al solicitante entregar los documentos requeridos —como condición de aprobación— y que el estudio se apruebe administrativamente sin necesidad de esperar a la próxima sesión de la Sala.
Que la Sala apruebe automáticamente (después de una notificación) cualquier estudio de investigación clínica que haya sido previamente aprobado por un ente regulador de un país de referencia (eje. Australia, Canadá, EE. UU., Unión Europea, Japón).
Que el acta que publica la Sala después de una sesión se publique en la página web del INVIMA máximo a los tres días hábiles después de la fecha de sesión de la Comisión Revisora.
Que el trámite de aprobación de un estudio de investigación clínica tome máximo 30 días hábiles y que se implemente el silencio administrativo (si el INVIMA no responde a la solicitud en 30 días, la aprobación será automática).
Que el proceso de aprobación local (CEI de la I.P.S. donde se realizará el estudio) sea en paralelo con el trámite de solicitud del concepto técnico del INVIMA.
Que la aprobación del registro de importación ante el VUCE sea automática (para esto el INVIMA debe de establecer un canal electrónico/digital de comunicación con el MinCIT).
Que los siguientes eventos en un estudio no requieran aprobación por el INVIMA sino que sean aprobados por el CEI de la I.P.S. donde se realizará el estudio y se le notifique al INVIMA en un tiempo prudente (eje. 30 días hábiles) después de su aprobación:
Cambio en el investigador principal
Adición de un nuevo centro de investigación
Actualizaciones en el manual del investigador
Cambios en el consentimiento informado
Cambios en el protocolo que no involucren cambios en el diseño o composición del producto médico en investigación
Que solo los siguientes eventos en un estudio requieran aprobación del INVIMA:
Cambios en el diseño del estudio que involucren cambios en el diseño o composición del producto médico en investigación
Cambio en las cantidades del producto médico en investigación a ser importado
Adición de nuevos productos a ser importados bajo el estudio
Cambio del titular de la autorización de importación y del registro de importación
Conclusión
Colombia tiene todo para convertirse en una potencia mundial en el desarrollo de estudios de investigación clínica como apoyo a la industria global de desarrollo de innovación médica. Colombia es consciente de que un crecimiento económico sostenible en el largo plazo no vendrá de la minería e hidrocarburos, sino de servicios sofisticados de alto valor agregado, manufactura e investigación y desarrollo. Este es el camino recorrido por casi todos los países de alto ingreso en los últimos 50 años. El país tiene en este campo un enorme potencial por cuenta, entre otras cosas, de la calidad de sus médicos, los buenos sistemas de información asociados al régimen de salud, la mejor infraestructura que ha logrado en los últimos años en laboratorios, clínicas y centros de diagnóstico, la ubicación geográfica y sus menores costos. Un trámite ágil en el INVIMA para la aprobación de estudios de investigación clínica contribuiría a generar hasta USD 500 millones anuales en exportación de servicios de salud si se atraen empresas extranjeras biofarmacéuticas y de dispositivos médicos para este propósito. Para lograrlo habrá que superar algunos obstáculos administrativos en el INVIMA que ayuden a reducir los tiempos de aprobación a 30 días para adelantar estudios de investigación clínica. El autor escribió otra columna titulada “Cómo Colombia en el 2020 podría recibir 100 estudios de investigación clínica anuales y exportar USD 500 millones en servicios de salud” que complementa esta columna; se le recomienda al lector leer ese otro artículo para un mejor entendimiento sobre el impacto socio-económico de crear un ambiente regulatorio ágil en Colombia para que se realicen más estudios de investigación clínica.
Acerca del autor
Julio G. Martinez-Clark es CEO de bioaccess.™ (www.bioaccessla.com); una empresa de investigación por contrato (CRO por sus siglas en inglés) que ayuda a fabricantes de dispositivos médicos a conducir estudios de investigación clínica en las I.P.S. de Colombia.